Cells with eosinophilic granular or oncocytic cytoplasm are a hallmark of benign renal oncocytoma, but they can be seen in other renal tumors. We report an unusual case of a large renal tumor with medium-sized oncocytic cells that were arranged in sheets and tubulocystic growth patterns. With histological, immunohistochemical and ultrastructural studies, a diagnosis of eosinophilic variant of chromophobe renal cell carcinoma (ChRCC) with tubulocystic features was made. The eosinophilic granular cytoplasm that was visible on the H&E sections corresponded to the abundant microvesicles packing the cytoplasm. Since the prognosis of ChRCC is regarded as more favorable than other subtypes of renal cell carcinomas (RCC), an accurate identification of this tumor has significant clinical implications. However, there is no current literature describing renal tumor similar to our case. Here we discuss the histologic features of this unique neoplasm at light and electron microscopic levels, the immunohistochemical profile, and differential diagnosis from its mimickers, including oncocytoma, clear cell RCC with granular cytoplasm and tubulocystic carcinoma of the kidney.
Authors: Yin, Ming ; Falls, Randall
Corresponding Author: Yin, Ming
Introduction
Chromophobe renal cell carcinoma (ChRCC) accounts for approximately 5% of all renal cell carcinomas (RCC).(1), (2), (3) Even though ChRCC is typically associated with a better prognosis than the other RCC subtypes; aggressive behavior has been seen in some cases.(2), (4) ChRCC is characterized by large pale cells with marked cell membranes resembling “plant cells”. However, accurate diagnosis of ChRCC can be challenging at times, especially when the tumor is purely composed of granular eosinophilic (oncocytic) cells. Histopathological studies on the eosinophilic variant of ChRCC have not been fully described in the literature, but this is possibly due to the fact that the histological findings of this variant of ChRCC are similar to those of oncocytoma. Histochemical and immunohistochemical stains may be helpful in some situations; however, so far, no markers can reliably distinguish between oncocytoma and ChRCC, particularly the eosinophilic variant.(5) We describe the histopathologic, immunohistochemical and ultrastructural findings of an eosinophilic variant of ChRCC with tubulocystic features, with emphasis on its problematic differential diagnosis from those conditions which mimic it.
Case Report
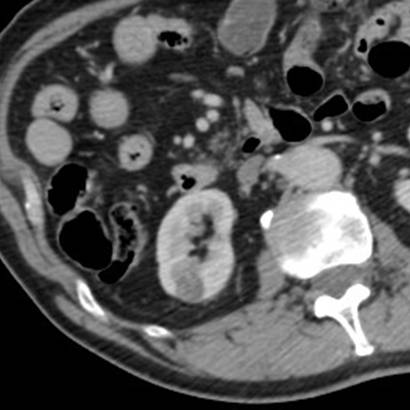
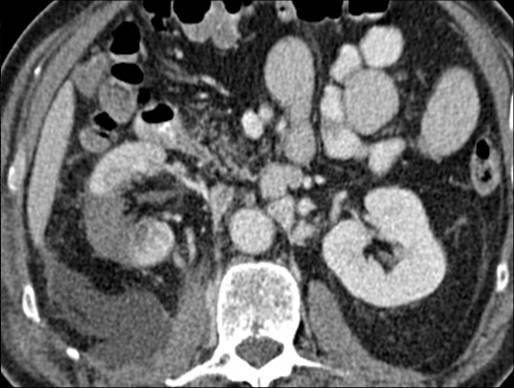
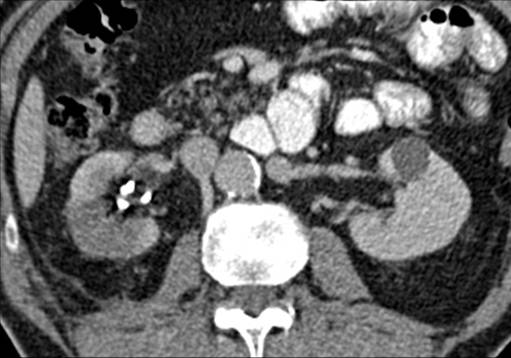
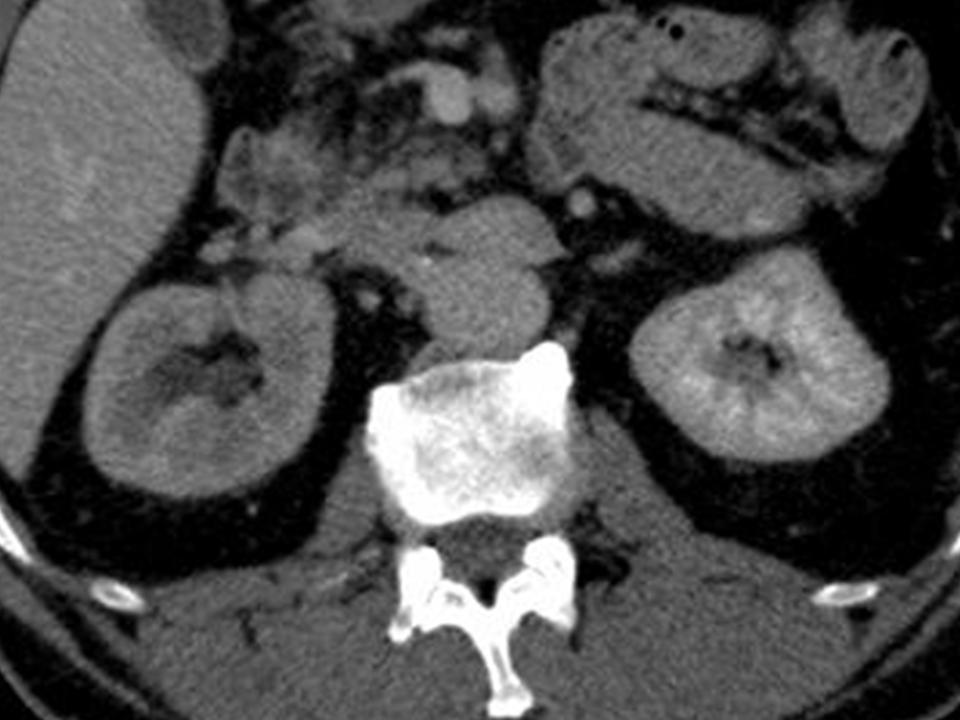
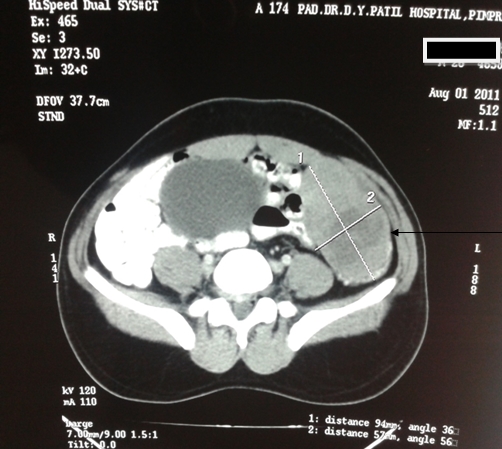
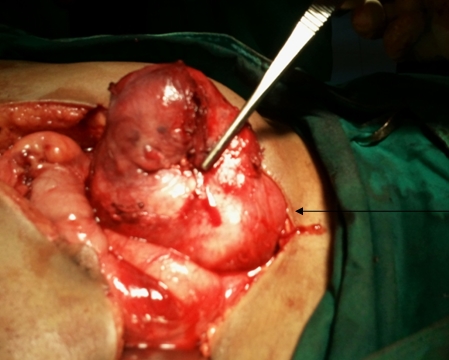
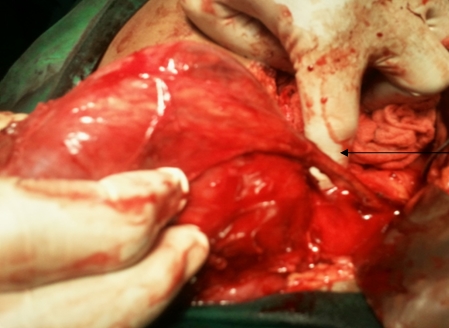
A 63-year-old female presented with a large left renal mass that was discovered during a work-up for abdominal pain of unknown duration. Computerized tomography of the abdomen revealed an exophytic, rounded mass arising from the upper pole of the left kidney with distortion of the renal collecting system. The patient subsequently underwent a left radical nephrectomy. The kidney specimen measured 16.8 x 10.0 x 6.8 cm and weighed 610 grams. On sectioning, the kidney showed a well-circumscribed tumor that measured 12.0 x 6.8 x 5.7 cm and occupied the upper pole, compressing the renal pelvis and calyceal system. The cut surface of the mass was homogeneously tan-pink in color with focal brown areas suggestive of old hemorrhage; no tumor necrosis or central scar was seen (Figure 1). No lymph nodes were noted.
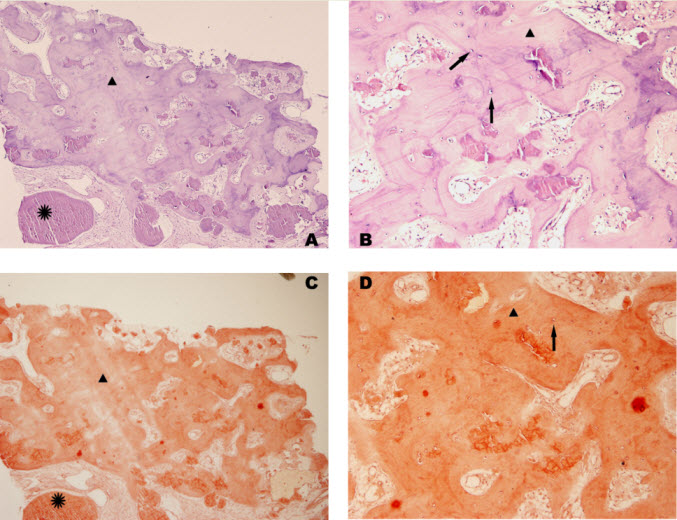
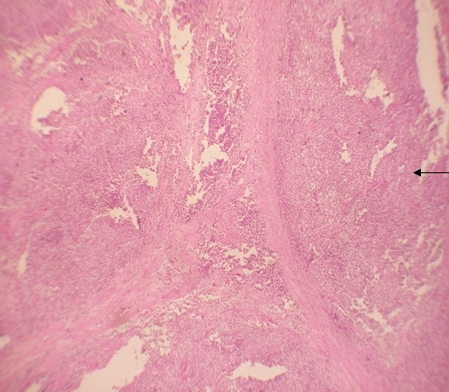
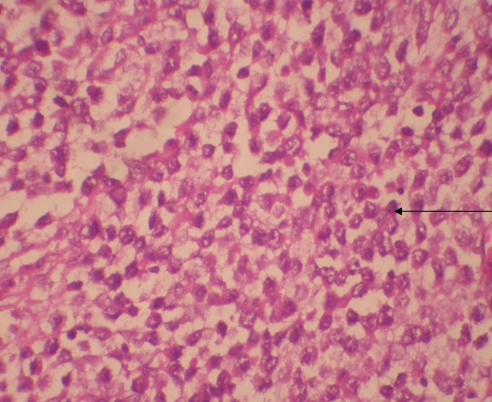
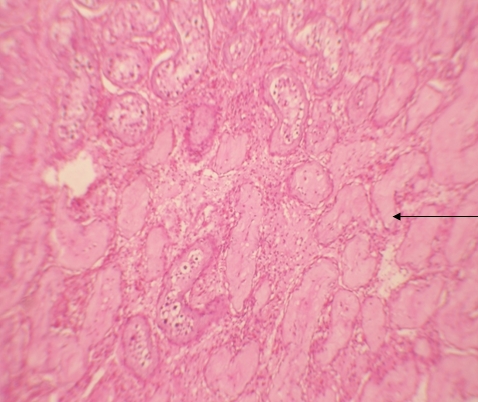
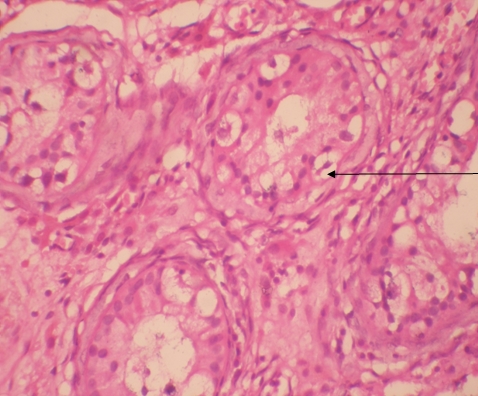
Histologic examination showed the mass to be composed of medium-sized polygonal cells, which displayed two architectural patterns. The majority of tumor cells were arranged in sheets or wide trabeculae (Figure 2). In adjacent areas, one could appreciate a tubulocystic growth pattern (Figure 3). The tumor cells had abundant granular eosinophilic cytoplasm with relatively well-defined cellular borders (Figure 4). There was mild to moderate nuclear pleomorphism, with an occasional thin-rim perinuclear halo. No sarcomatoid transformation was seen. The immunohistochemical stains showed diffuse immunoreactivity for pancytokeratin (AE1/3), cytokeratin 7, Ber-EP4, and CD117 (Figure 5). The stains with antibodies against RCC marker, CD10 and vimentin were negative. There was also cytoplasmic positive staining with Hale’s colloidal iron. Electron microscopy showed that abundant microvesicles, admixed with mitochondria, were distributed diffusely throughout the cytoplasm (Figure 6). On higher power, the microvesicles were bound by smooth single membranes, measuring 200 – 500 nm in diameter, and packing the spaces between mitochondria (Figure 7). A small number of lysosomes, lipid vacuoles and glycogen granules were noted in the cytoplasm, where the Golgi apparatus and the rough endoplasmic reticulum were poorly developed. Nuclei were rounded with irregular folded nuclear membrane and heterochromatin.
Discussion
When renal tumor cells show distinctive granular eosinophilic cytoplasm, the main diagnostic challenge is to separate an eosinophilic variant of ChRCC from the benign renal oncocytoma. Grossly, 33% of large oncocytomas may have a central stellate scar, which was not visible in the current tumor. Histologically, oncocytoma usually has tumor cells with inconspicuous cell borders arranged in nests or acini in a hypocellular hyalinized stroma, features that were not seen in this tumor either. Nuclear membrane irregularity and coarse chromatin are noted in this case, while oncocytomas usually have cells showing bland, uniform nuclei with smooth nuclear membranes. Hale’s colloidal iron stain showed diffuse cytoplasmic positivity in our case, whereas oncocytoma often displays focal positive staining confined to the luminal borders. Immunohistochemical stains performed on the current tumor showed that the cells were positive for cytokeratin 7, Ber-EP4, and CD117; and negative for RCC marker, CD10 and vimentin; a staining profile favoring a ChRCC. However, there is no established panel of immunohistochemical stains that can differentiate ChRCC from oncocytoma with certainty. This could, in part, be explained by the fact that these two tumors may share a common cell of origin in the distal nephron, namely intercalated cells of the collecting ducts.(5), (6) Electron microscopic analysis is considered the only definitive method to consistently distinguish these two entities.(7) The granular appearance of the cytoplasm on light microscopy corresponds to the ultrastructural finding of abundant microvesicles and scattered mitochondria in the cytoplasm of a majority of tumor cells, which is considered diagnostic for ChRCC.(7) In contrast; it is known that oncocytoma is characterized by the packing of the cytoplasm by mitochondria with paucity of microvesicles and other cell organelles. Unlike the classic ChRCC, the cytoplasm of this tumor was filled with crowded microvesicles diffusely interspersed between mitochondria, leading to less prominence or absence of perinuclear clearing (halo) by light microscopy. Previous studies have suggested that the microvesicles are derived from mitochondria because of the close relationship between microvesicles and mitochondria at the ultrastructural level;(8) although the possibility of endoplasmic reticulum being the origin has also been suggested.(1), (7) In addition, clear cell RCC can occasionally show a somewhat granular cytoplasm leading to potential confusion with ChRCC.(4), (7) However, clear cell RCC usually demonstrates alveolar or acinar growth patterns with delicate thin-walled blood vessels by light microscopy and abundant cytoplasmic lipid vacuoles and glycogen granules by electron microscopy. Immunohistochemically and histochemically, clear cell RCC is usually positive for antibodies against vimentin, RCC marker and CD10, and negative for CD117 and colloidal iron.
ChRCC was first described in 1985 as a tumor composed of cells with unique histomorphologic and ultrastructural features.(1) The tumor is presumably derived from intercalated cells of the collecting ducts.(5) Histologically, ChRCC is characterized by polygonal cells with distinct nuclear borders and evenly distributed, finely granular, translucent and pale cytoplasm. The classic architectural appearance is that of sheets or wide trabeculae of cells often separated by incomplete fibrovascular septations. Occasional, papillary, tubulocystic and nested patterns may be present.(1), (4) The nuclei often are hyperchromatic with nuclear membrane irregularity (raisinoid) and coarse chromatin. Binucleation or multinucleation are also common. Classically, perinuclear cytoplasmic clearing (halo) is present. Interestingly, it appears that nuclear grade as determined by the Fuhrman system may not reflect the clinical or biological behavior of chromophobe RCC as closely as it does for the other subtypes.(9)
The eosinophilic variant of ChRCC was first reported in 1997.(10) It usually shows small to medium-sized, eosinophilic granular/oncocytic cells arranged in sheets.(11) Compared with the classic ChRCC, our case showed smaller cell size, less prominent cell borders and thinner or absent perinuclear halos. However, similar to classic ChRCC, the eosinophilic variant is a tumor of low malignant potential with reported 5-year and 10-year survival rates of 78-100% and 80-90%, respectively.(1), (11) It is also believed that the cytogenetic abnormalities of this variant are similar to those of the classic ChRCC, including losses of chromosomes 1, 2, 6, 10, 13, 17 and 21.(12), (13)
ChRCC with microcystic and adenomatous patterns was first described in 1998.(14) Because of the tubulocystic architectures identified in the current case, a differential diagnosis that includes tubulocystic carcinoma of the kidney is needed. The tubulocystic carcinoma of the kidney is a recently recognized subtype of renal tumor that is not listed in the 2004 WHO classification.(3) Besides the packed tubules and cysts separated by bland fibrous stroma, the lining tumor cells of this newly described entity are cuboidal to columnar with abundant eosinophilic cytoplasm and usually large nuclei with prominent nucleoli.(15) These features are readily separable from the current case by routine histology. In addition, the genetic signatures of the tubulocystic carcinoma of the kidney are gains of chromosome 7 and 17.
In conclusion, we report for the first time a unique case of an eosinophilic variant of ChRCC that contained tubulocystic features. Because ChRCC is associated with a different prognosis from those conditions which mimic it, accurate identification of this tumor has important clinical implications. Even if the cytological features of this eosinophilic variant of ChRCC are extremely similar to that of renal oncocytoma, we believe that the combination of careful evaluation of the architectural patterns, cytologic characteristics and special stain profile of adequately sampled tumor will be very helpful for its accurate identification. If needed, ultrastructural study will yield a definitive diagnosis. Likewise, the unique morphologic and architectural patterns will help to distinguish the eosinophilic variant of ChRCC from clear cell RCC with granular cells and tubulocystic carcinoma of the kidney. Further elucidation of the characteristic cytogenetic abnormalities will provide a foundation for molecular classification of the neoplasm.
Acknowledgement
The authors thank Dr. Peter Kragel for his advices and critical comments on the manuscript and Debra Laich for her expert help in electron microscopy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
(1) Thoenes W, Storkel S, Rumpelt HJ. Human chromophobe cell renal carcinoma. Virchows Arch B Cell Pathol Incl Mol Pathol 1985;48(3):207-217.
(2) Przybycin CG, Cronin AM, Darvishian F, Gopalan A, Al-Ahmadie HA, Fine SW, et al. Chromophobe renal cell carcinoma: a clinicopathologic study of 203 tumors in 200 patients with primary resection at a single institution. Am J Surg Pathol 2011 Jul;35(7):962-970.
(3) Eble JN, Sauter G, Epstein JI, Sesterhenn IA. World Health Organization Classification of Tumours. Pathology and genetics of tumors of the urinary system and male genital organs. Lyon: IARC Press; 2004.
(4) Amin MB, Paner GP, Alvarado-Cabrero I, Young AN, Stricker HJ, Lyles RH, et al. Chromophobe renal cell carcinoma: histomorphologic characteristics and evaluation of conventional pathologic prognostic parameters in 145 cases. Am J Surg Pathol 2008 Dec;32(12):1822-1834.
(5) Truong LD, Shen SS. Immunohistochemical diagnosis of renal neoplasms. Arch Pathol Lab Med 2011 Jan;135(1):92-109.
(6) Ortmann M, Vierbuchen M, Fischer R. Sialylated glycoconjugates in chromophobe cell renal carcinoma compared with other renal cell tumors. Indication of its development from the collecting duct epithelium. Virchows Arch B Cell Pathol Incl Mol Pathol 1991;61(2):123-132.
(7) Tickoo SK, Lee MW, Eble JN, Amin M, Christopherson T, Zarbo RJ, et al. Ultrastructural observations on mitochondria and microvesicles in renal oncocytoma, chromophobe renal cell carcinoma, and eosinophilic variant of conventional (clear cell) renal cell carcinoma. Am J Surg Pathol 2000 Sep;24(9):1247-1256.
(8) Moreno SM, Benitez IA, Martinez Gonzalez MA. Ultrastructural studies in a series of 18 cases of chromophobe renal cell carcinoma. Ultrastruct Pathol 2005 Sep-Oct;29(5):377-387.
(9) Paner GP, Amin MB, Alvarado-Cabrero I, Young AN, Stricker HJ, Moch H, et al. A novel tumor grading scheme for chromophobe renal cell carcinoma: prognostic utility and comparison with Fuhrman nuclear grade. Am J Surg Pathol 2010 Sep;34(9):1233-1240.
(10) Erlandson RA, Shek TW, Reuter VE. Diagnostic significance of mitochondria in four types of renal epithelial neoplasms: an ultrastructural study of 60 tumors. Ultrastruct Pathol 1997 Sep-Oct;21(5):409-417.
(11) Manipadam MT, Korula A, Chandrasingh J, Devasia A. Chromophobe renal cell carcinoma: A report of two cases with unusual histological features. Indian J Urol 2008 Jan;24(1):123-125.
(12) Brunelli M, Eble JN, Zhang S, Martignoni G, Delahunt B, Cheng L. Eosinophilic and classic chromophobe renal cell carcinomas have similar frequent losses of multiple chromosomes from among chromosomes 1, 2, 6, 10, and 17, and this pattern of genetic abnormality is not present in renal oncocytoma. Mod Pathol 2005 Feb;18(2):161-169.
(13) Yusenko MV. Molecular pathology of chromophobe renal cell carcinoma: a review. Int J Urol 2010 Jul;17(7):592-600.
(14) Michal M, Hes O, Svec A, Ludvikova M. Pigmented microcystic chromophobe cell carcinoma: a unique variant of renal cell carcinoma. Ann Diagn Pathol 1998 Jun;2(3):149-153.
(15) Azoulay S, Vieillefond A, Paraf F, Pasquier D, Cussenot O, Callard P, et al. Tubulocystic carcinoma of the kidney: a new entity among renal tumors. Virchows Arch 2007 Nov;451(5):905-909.
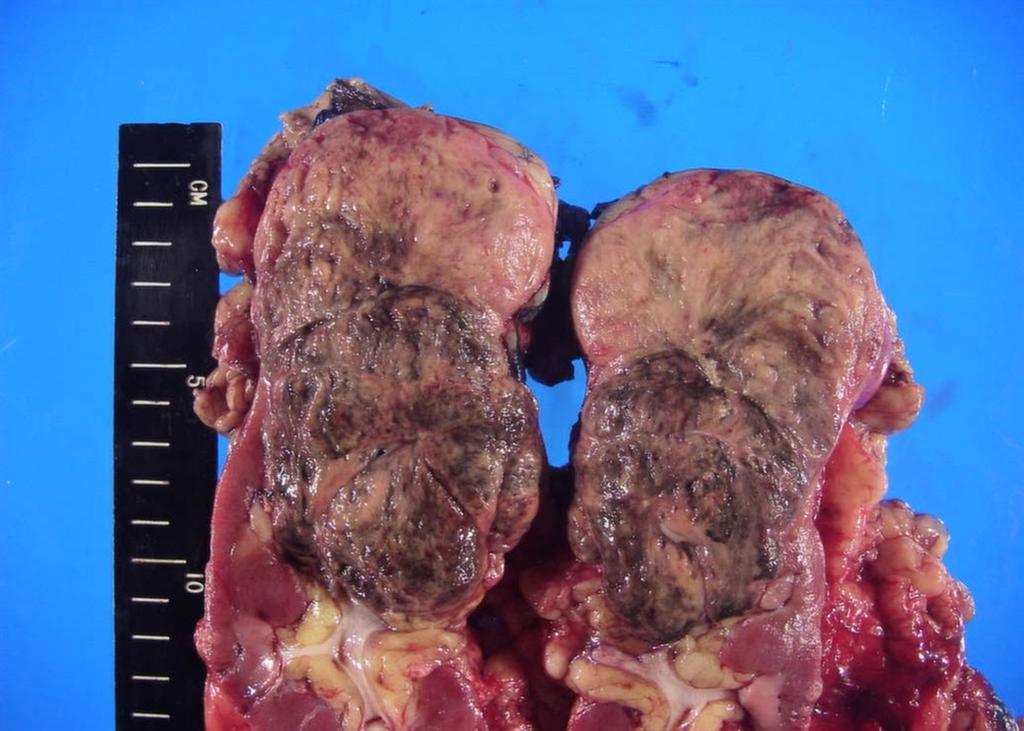
Figure 1. The upper pole mass was homogeneously tan-pink in color with dark brown areas suggestive of old hemorrhage.
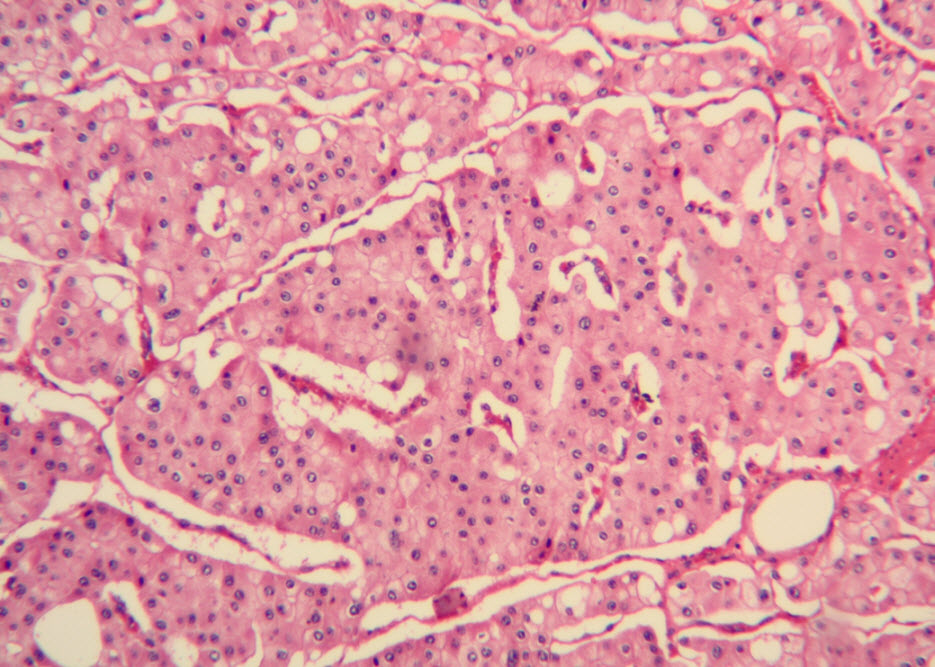
Figure 2. The majority of tumor cells were arranged in sheets or wide trabeculae. (hematoxylin and eosin, 100x)
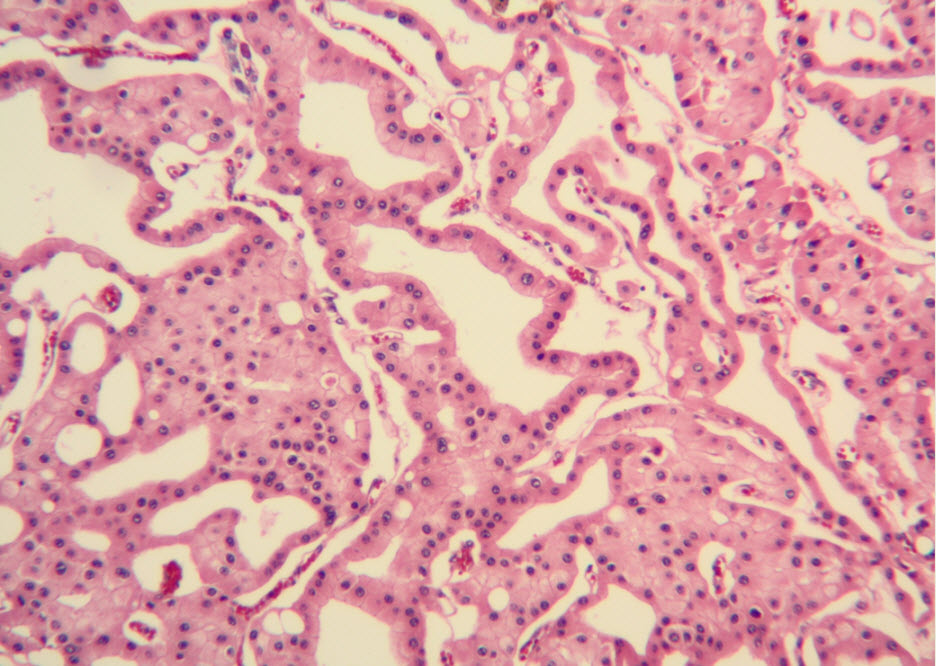
Figure 3. Adjacent to wide trabeculae, tumor cells show tubulocystic growth pattern. (hematoxylin and eosin, 100x)
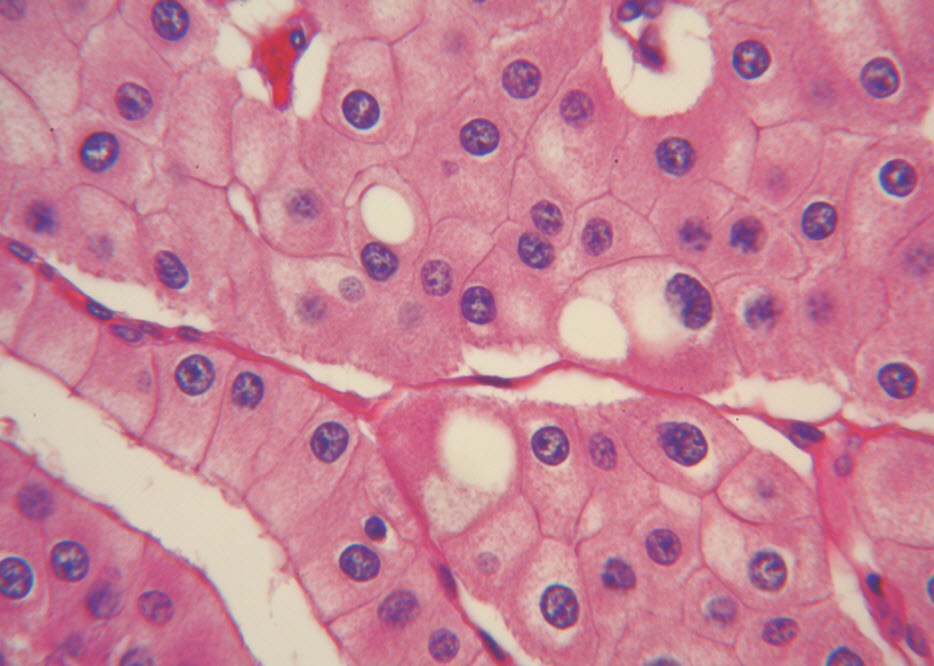
Figure 4. The polygonal tumor cells have abundant granular eosinophilic cytoplasm with relatively welldefined cellular borders. There was mild to moderate nuclear pleomorphism, with occasional thin-rim perinuclear halo. (hematoxylin and eosin, 400x )
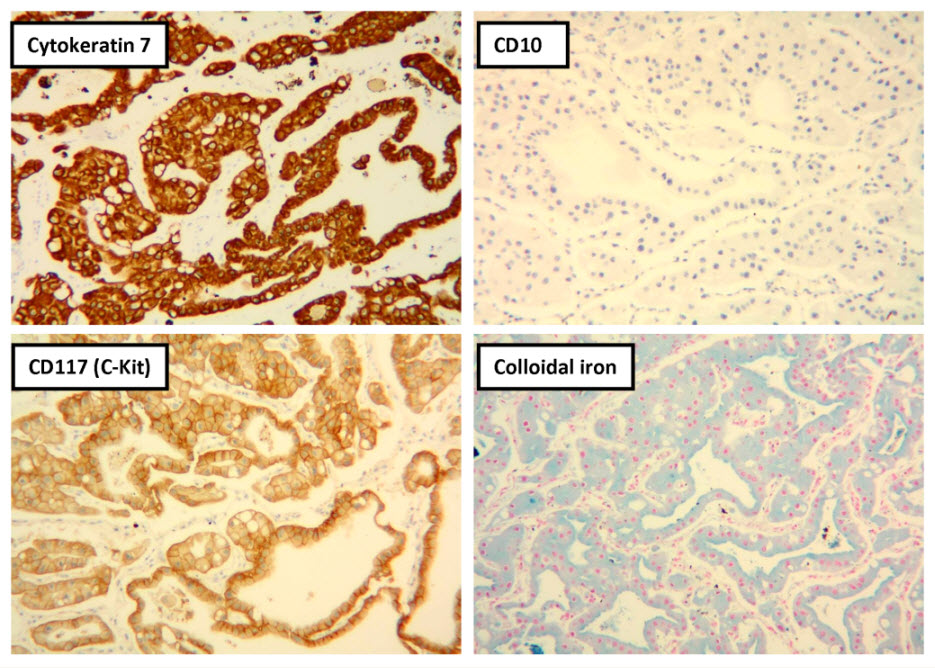
Figure 5. Selected photomicrographs of the renal mass with immunohistochemical and Hale’s colloidal iron stains are shown. (x100)
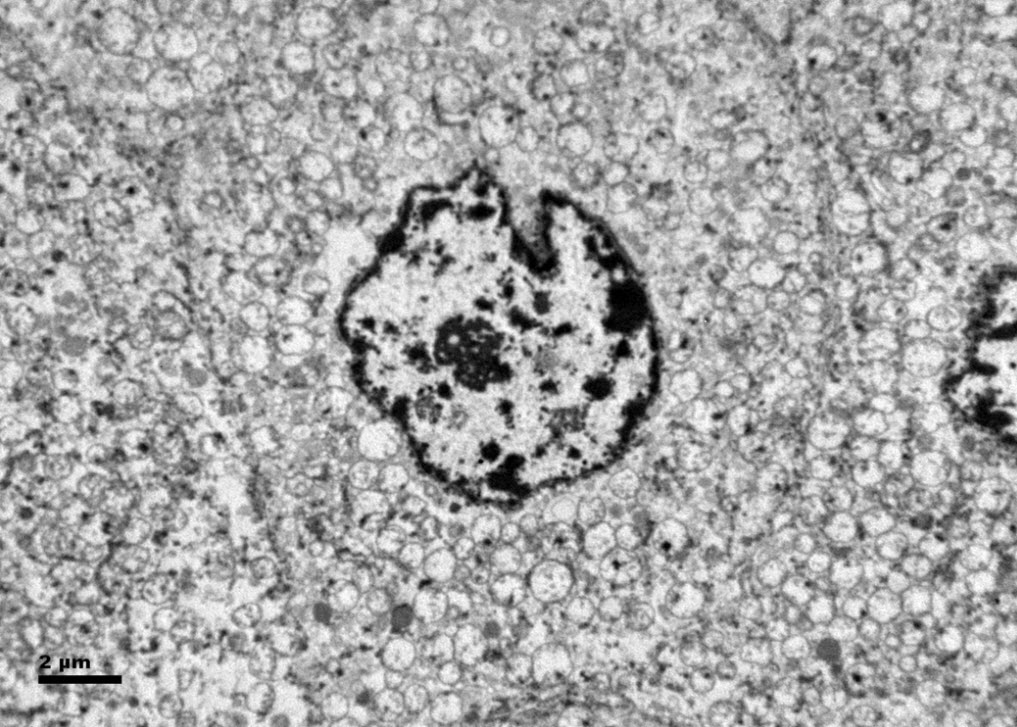
Figure 6. Ultrastructurally, the tumor cells show abundant microvesicles, admixed with mitochondria, distributed diffusely throughout the cytoplasm. Nuclei were rounded with irregular nuclear membranes and coarse chromatin.
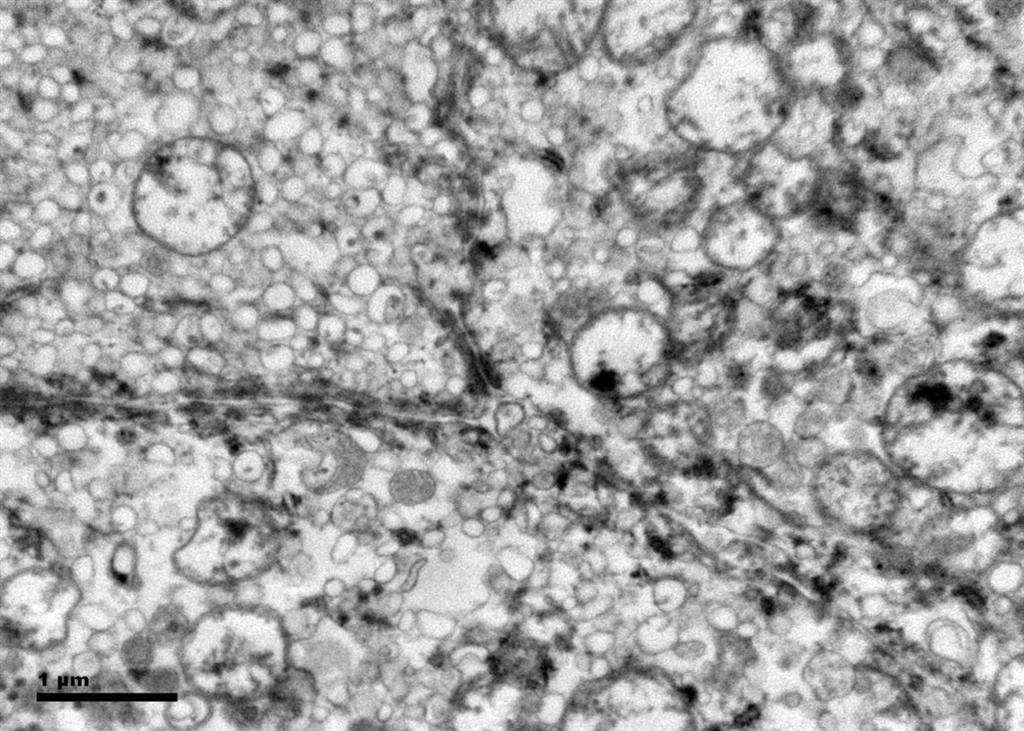
Figure 7. The crowded microvesicles show smooth single membranes, measure 200 – 500 nm in diameter, and pack the spaces between mitochondria. A small number of lysosomes, lipid vacuoles and glycogen granules are also present.
Date added to bjui.org: 06/11/2012
DOI: 10.1002/BJUIw-2012-048-web