
Metastatic Colon Adenocarcinoma to the Tunica Vaginalis Testis Presenting As Hydrocele
To the best of our knowledge, ours is the first case of tunica vaginalis metastasis originating from a primary adenocarcinoma of the ascending colon.
Authors: Ji Zheng1, Gensheng Lu1, Chengping Xu2, Bo Song1, Zhansong Zhou1
1. Department of Urology, Southwest hospital, Third Military Medical University, Chongqing, China
2. Department of Pathology, Southwest hospital, Third Military Medical University Chongqing, China
Corresponding Author: Zhansong Zhou, Department of Urology, Southwest hospital, Third Military Medical University, Chongqing, China. E-mail: [email protected]
Case report
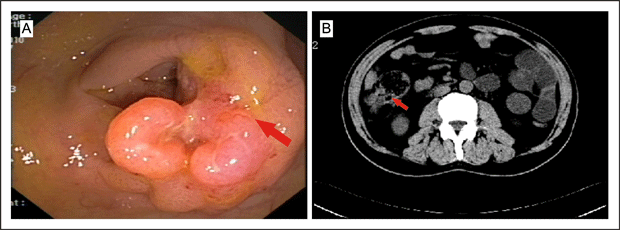
A 65-year-old man was referred to our urology clinic complaining of left scrotal enlargement for one month. Seven months before admission, colonoscopy (Figure 1A) and computed tomography (CT) of his abdomen (Figure 1B) showed a mass in his ascending colon and enlarged mesenteric lymph nodes.
Figure 1A and B. CT scan of the abdomen
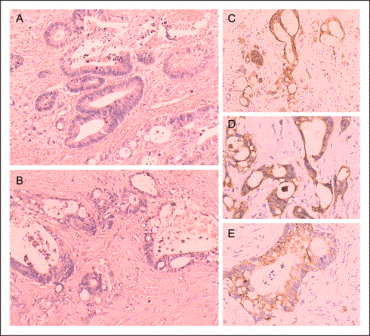
Biopsy was consistent with a moderately differentiated adenocarcinoma. He was diagnosed preoperatively with ascending colonic adenocarcinoma, and mesenteric lymph nodes metastases were suspected. Due to his experiencing severe intestinal spasm and bloody stools, the patient requested surgical treatment, though systematic chemotherapy had been advised. He underwent a radical right hemicolectomy, and this pathology revealed moderately differentiated adenocarcinoma with lymph node metastases (Figure 3A).
Figure 3A. Pathology revealed moderately differentiated adenocarcinoma with lymph node metastases
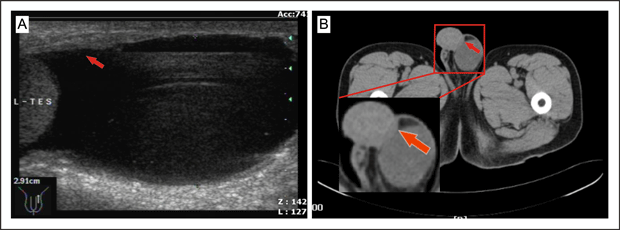
Two weeks after surgery, in view of his having lymphatic metastases, our patient agreed to and completed the first cycle of a combined chemotherapy regimen with oxaliplatin (150 mg intravenous [IV] day 2), tegafur (600 mg IV days 1 to 3), and calcium folinate (400 mg IV days 1 to 3). Another four cycles of combined chemotherapy were accomplished in the following four months. One month before referral to urology, an ultrasound examination had shown a hydrocele of the left tunica vaginalis testis, thickening of the left perididymis, with a normal appearance of the testis and epididymis (Figure 2A). Ten days previously, a painless intrascrotal nodule had been palpated accidentally and scrotal examination revealed a hard flat mass, 1.5×0.6×0.3 cm in diameter, adhering to the left tunica vaginalis testis. CT scan demonstrated a mass in this area (Figure 2B).
Figure 2A and B. CT scan
Serum levels of the tumor-associated proteins CA242 and CA19-9 were significantly elevated to 268 kU/L (normal range 0-20 kU/L) and 372 kU/mL (normal range 0-35 kU/mL), respectively, while those of cancer antigens, carcinoembryonic antigen, neuron-specific enolase, alpha-fetoprotein, prostate specific antigen, CA125, CA15-3 and beta human chorionic gonadotropin were within their normal ranges.
The patient underwent left orchiectomy through an inguinal approach and the mass was found to be densely fixed to the left tunica vaginalis testis but separated from the testis, epididymis and spermatic cord. Frozen sections suggested the mass to be an adenocarcinoma. Interestingly, a left oblique inguinal hernia with greater omentum inside was found and the greater omentum was in direct contact with the lesion of the tunica vaginalis testis. A hernia repair was performed accordingly.
Subsequent biopsy revealed a moderately-differentiated adenocarcinoma infiltrating the tunica vaginalis testis without involvement of the testis or epididymis (Figure 3B). The primary colonic tumor and this tumor shared identical neoplastic glands with intra-cytoplasmic mucin and moderate nuclear pleomorphism (Figure 3A, 3B). Immunohistochemistry was performed using the immunoperoxidase method. The tumor cells showed positivity for Villin (Figure 3C), CK(Figure 3D) and CK20(Figure 3E), but negative for D2-40, TTF1, WT1, Ki67, CK7, SMA, and PSA, which suggested an occult gastrointestinal or colorectal origin of the carcinoma, with a lesser probability of carcinoma of the lung, liver, thyroid, breast, prostate, or hematological system.[1,2]
Discussion
Solid tumor metastasis to the testis and paratesticular region is rare and principally encountered as an incidental autopsy finding.[3] Metastatic carcinoma accounted for respectively five of 112 and nine of 66 paratesticular tumors reviewed.[4,5] The common primary sites of metastasis of the paratesticular tissue are the prostate, kidney, gastrointestinal tract, lung, and breast.[6]
Tumors of the tunica vaginalis testis, both primary and secondary, and whether benign or malignant, are extremely rare.[7,8] Fewer than 10 cases, including our case, have been reported to date in the English and non-English literature,[8–13] and the primary sites for metastatic tumor of tunica vaginalis testis include stomach,[8–10,12] rectum,[11] cecum ,[12] and colon (as in our case). Most cases present with paratesticular mass or hydrocele, and they may or may not be associated with pain.
Metastasis to the paratesticular tissues can be the first sign of tumor recurrence and more rarely present as the first manifestation of an occult primary neoplasm.[14] Our review revealed hydrocele of the tunica vaginalis testis presented as the first clinical manifestation of primary [9] and secondary [8,10–12] and our case tunica vaginalis testis tumors, which suggested that cancerous lesions should be considered in the management of hydrocele, especially in the follow-up of cancer patients. CT or PET-CT is suggested as a rapid and reliable modality for facilitating a preoperative diagnosis.
Clinically, most patients with metastatic paratesticular tumors suffer from a painless intrascrotal mass and the diagnosis is frequently delayed. Hydrocele, inguinal hernia, epididymal cyst, spermatocele and spermatic cord tumors are the most common misdiagnoses of the tumor.[15] The consideration of these diseases in the differential diagnosis of a painless paratesticular mass is critical, as it may allow more precise diagnosis and more targeted treatment, especially in high-risk patients.
Infiltrative lesions of the epididymis [9] and spermatic cord [10,11] can be found in some metastatic tunica vaginalis testis tumor cases, which implies that other primary sites of tunica vaginalis testis metastases may be similiar to primary sites of paratesticular metastatic tumors, and include prostate, gastrointestinal tract, kidney, lung and breast.[15, 16]
Proposed routes of metastasis to the tunica vaginalis testis may include direct extension from adjacent organs, intraductal spread via the vas deferens, retrograde venous and lymphatic extension, arterial embolism [9], and transperitoneal seeding through a patent tunica vaginalis [17] or congenital hydrocele [18]. In our case, the possibility of lymphatic or hematogenous spread was suspected, particularly in view of the primary tumor with lymph node metastasis. However, the presence of a left oblique inguinal hernia in direct contact with the lesion of the tunica vaginalis testis made local tissue invasion another possible mode of spread .[19]
In our review, the median age of patients with metastatic tumor of the tunica vaginalis testis is approximately 62 years, with a reported age range from 50 to 67 years. Tumors metastasising to the paratesticular tissue usually are identified in the setting of disseminated disease and occur only uncommonly as a solitary site of metastasis.[2] The prognosis generally is poor, with the average survival period from the time of diagnosis of the metastasis being 9.1 months.[2] Chemotherapy is the only possible option for the treatment of a secondary tumor at the distant metastatic stage. Our patient completed four cycles of a combined chemotherapy regimen with oxaliplatin (120 mg intravenous [IV] day 1), fluorouracil (500 mg IV days 1 to 2), and calcium folinate (200 mg IV days 1 to 2). He is alive and has been under careful surveillance for about five months up to the time of this report.
In our case and in others reviewed, once paratesticular metastasis had occurred, disease control or cure was negligible.[20] Whether or not a more aggressive approach would lead to a different outcome remains unknown. In some patients, early diagnosis by cytological examination of hydrocele fluid [21] or ultrasound-guided fine-needle aspiration cytology [22] may be helpful for diagnosis and prognosis, as well as treatment of secondary tumors of the tunica vaginalis testis.
To the best of our knowledge, ours is the first case of tunica vaginalis metastasis originating from a primary adenocarcinoma of the ascending colon. Though rare, metastatic lesions of the tunica vaginalis testis should be included in the differential diagnosis of a patient with ambiguous paratesticular masses or in the setting of a hydrocoele. High awareness of this entity may lead to earlier diagnosis and more timely and appropriate treatment, despite the unfavorable prognosis for treatment of secondary tumor of tunica vaginalis testis at present.
References
1.Park SY, Kim HS, Hong EK, et al: Expression of cytokeratins 7 and 20 in primary carcinomas of the stomach and colorectum and their value in the differential diagnosis of metastatic carcinomas to the ovary. Hum Pathol 33:1078-1085, 2002
2.De Lott LB, Morrison C, Suster S, et al:CDX2 is a useful marker of intestinal-type differentiation: a tissue microarray-based study of 629 tumors from various sites. Arch Pathol Lab Med 129:1100-1105, 2005
3.Meacham RB, Mata JA, Espada R,et al:Testicular metastasis as the first manifestation of colon carcinoma. J Urol 116:621-622, 1988
4.Lioe TF, Biggart JD: Tumours of the spermatic cord and paratesticular tissue. A clinicopathological study. Br J Urol 71:600-606, 1993
5.Hartwick RW, Srigley JR, Burns B, et al: A clinicopathologic review of 112 paratesticular tumours[Abstract]. Laboratory Invest 56:83, 1987
6.Algaba F, Santaularia JM, Villavicencio H: Metastatic tumor of the epididymis and spermatic cord. Eur Urol 9:56-59, 1983
7.Plas E, Riedl CR, Pflüger H: Malignant mesothelioma of the tunica vaginalis testis: review of the literature and assessment of prognostic parameters. Cancer 83:2437-2446, 1998
8.Lee J, Kang SC, Ban JH, et al: Metastatic Tumor of Tunica Vaginalis Testis with Hydrocele in a Patient with Gastric Cancer. Korean J Urol 48:667-669, 2007
9.Yeo JK:Scrotal Hydrocele as the First Clinical Manifestation of Occult Gastric Cancer.Korean. J Urol 50:1151-1153, 2009
10.Kageyama Y, Kawakami S, Li G, et al: Metastatic tumor of spermatic cord and tunica vaginalis testis from gastric cancer: a case report . Hinyokika Kiyo 43:429-431, 1997
11.Yasuhide T, Heizo Y, Katsusuke N: Metastatic Tumor of the Tunica Vaginalis Testis Arising from Rectal Cancer:A Case Report. Nishinihon Journal of Urology 61:245-247, 1999
12.Wai HP, Yau TK, Sze WM, et al:Metastatic tumour of the tunica vaginalis testis from carcinoma of the stomach. Int J Clin Pract 54:685-686, 2000
13.Ruiz JM, Sanchis CM, López PC, et al: Metastasis to the tunica vaginalis testis from a primary mucinous tumor of the cecum. Arch Esp Urol 63:235-238, 2010
14.Bennett VS, Bailey DM: Cholangiocarcinoma presenting as a solitary epididymal metastasis: a case report and review of the literature. Diagn Pathol 2:33, 2007
15.Beccia DJ, Krane RJ, Olsson CA. Clinical management of non-testicular intrascrotal tumors. J Urol 116:476-479, 1976
16.Algaba F, Santaularia JM, Villavicencio H: Metastatic tumor of the epididymis and spermatic cord. Eur Urol 9:56-59, 1983
17.Dutt N, Bates AW, Baithun SI: Secondary neoplasms of the male genital tract with different patterns of involvement in adults and children. Histopathology 37:323-331, 2000
18.Hanash KA, Carney JA, and Kelalis PP: Metastatic tumors to the testicles:routes of metastasis. J Urol 102: 465-468, 1969
19.Galanis I, Chatzimavroudis G, Katsougiannopoulos A, et al: Spermatic cord metastasis presenting as strangulated inguinal hernia – first manifestation of a multifocal colon adenocarcinoma: a case report. Cases J 2:61, 2009
20.Charles W, Joseph G, Hunis B, et al: Metastatic colon cancer to the testicle presenting as testicular hydrocele. J Clin Oncol 23:5256-5257, 2005
21.Gupta SC, Gupta AK, Misra V, et al: Pre-operative diagnosis of malignant mesothelioma of tunica vaginalis testis by hydrocele fluid cytology. Eur J Surg Oncol 24:153-154, 1998
22.Bruno C, Minniti S, Procacci C:Diagnosis of malignant mesothelioma of the tunica vaginalis testis by ultrasound-guided fine-needle aspiration. J Clin Ultrasound 30:181-183, 2002
Date added to bjui.org: 19/02/2012
DOI: 10.1002/BJUIw-2011-067-web




















